×
![]()
Greetings!
I am a postdoctoral researcher living in the San Francisco Bay Area, originally from the greater Philadelphia area, with a deep interest in the intersection of computational chemistry, machine learning, and statistical physics. My research focuses on improving molecular models by integrating various sources of information with Bayesian inference, maximum entropy methods, and deep learning techniques to bridge the gap between theory and experiment. These approaches have applications in biophysics, structural biology, and drug discovery, where accurate predictive models are essential for understanding complex molecular systems.
Currently, I am a member of Andrej Šali's group and Rada Savic's group. My work centers on method development for metamodeling — an integrative framework for combining models operating at different spatial and temporal scales with experimental data to construct a unified, data-driven representation of complex biological systems. We are using this approach to study the action of tuberculosis drugs, with the broader goal of supporting collaborative efforts to integrate individual models of cellular processes. This involves connecting molecular models (structural biology), network models (systems biology), and compartmental models (PK/PD) into a cohesive, multiscale representation of drug action.
I received my Ph.D. in Theoretical/Physical Chemistry from Temple University under the fantastic mentorship of Dr. Vincent A. Voelz. During my time in the Voelz lab, my primary focus was to develop and apply Bayesian inference methods that enhance forward models, force field parameterization, ensemble refinement, and predictive modeling of biomolecular systems. In other projects, I created and analyzed large-scale all-atom molecular dynamics simulations to study the thermodynamics and kinetics of protein folding and protein-protein binding systems.
Beyond research, I love my beautiful wife, hanging out with family and friends, being outdoors, playing sports, exercising, and experiencing diverse cultures while meeting new people.
More information about my research, publications, and projects can be found on this website. Feel free to reach out if you’re interested in collaborations or discussing cool science!
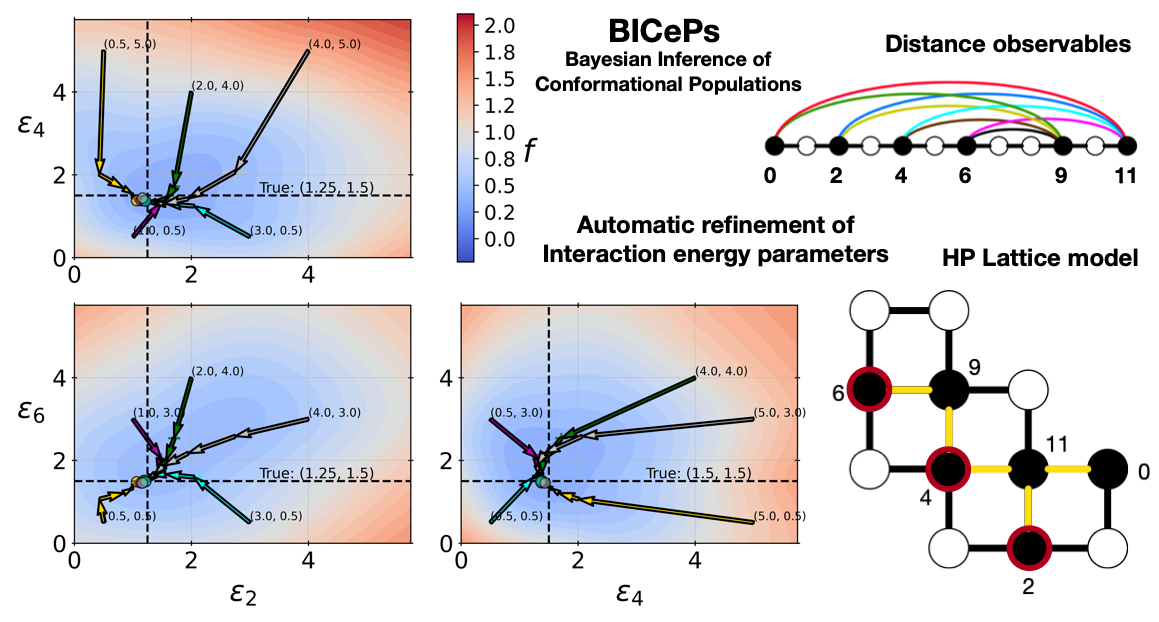
How can we properly refine our physical models using ensemble-averaged experimental data (e.g., NMR, SAXS, Cryo-EM, etc.) while accounting for error in the data? Can we variationally minimize our free energy-like quanitity (the BICePs score) to optimize force field parameters?
Robert M. Raddi and Vincent A. Voelz. "Automated optimization of force field parameters against ensemble-averaged measurements with Bayesian Inference of Conformational Populations." arxiv.org/abs/2402.11169
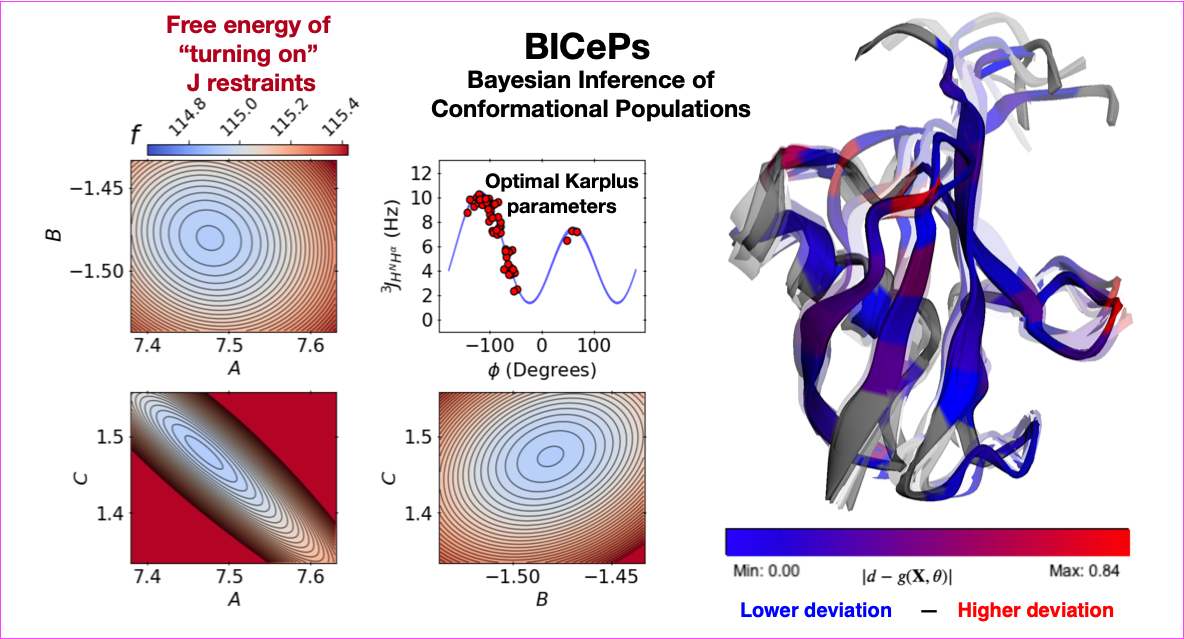
Forward models enable us to make predictions of experimental observables from simulated structures. Many forward models are empirical and require fine-tuning for particular systems we wish to study e.g., the Karplus relation. How can we best determine Karplus parameters without having a crystal structure? Here, we demonstrate our algorithm's ability to perform joint forward model optimization across six sets of Karplus coefficients. Furthermore, we show how this approach naturally generalizes to any differentiable forward model, such as those constructed with neural networks.
Robert M. Raddi, Tim Marshall and Vincent A. Voelz. "Automatic Forward Model Parameterization with Bayesian Inference of Conformational Populations." APL Mach. Learn. 4, 016102 (2026), preprint; article
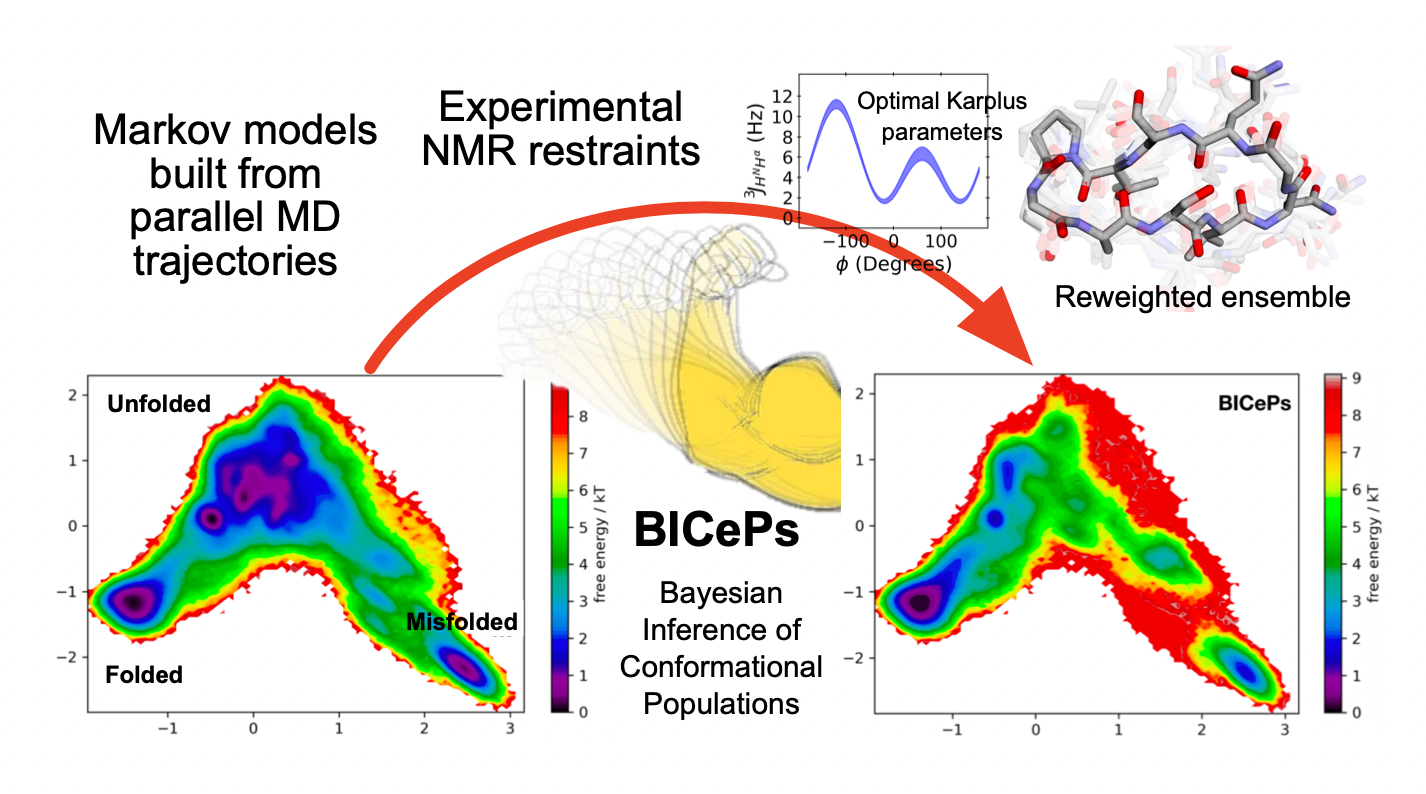
Will the inclusion of experimental data restraints improve predicitons of folding stability? Does finding generalized Karplus parameters across these twelve systems help our predictions?
Thi Dung Nguyen$^{\large\boldsymbol{\dagger}}$, Robert M. Raddi$^{\large\boldsymbol{\dagger}}$, and Vincent A. Voelz. "High-resolution tuning of non-natural and cyclic peptide folding landscapes against NMR measurements using Markov models and Bayesian Inference of Conformational Populations."J. Chem. Theory Comput. (2025), preprint; article
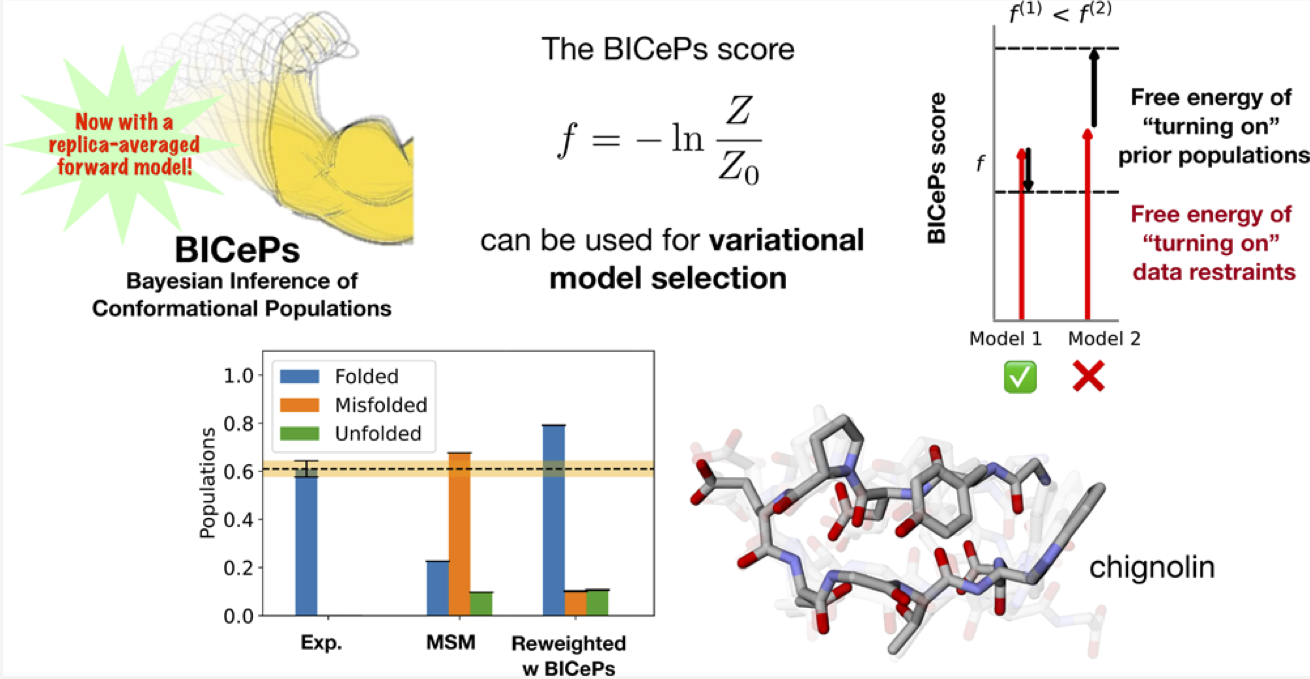
How do we know our physical models (e.g., force fields) are good? What is the best way to objectively rank them?
Robert M. Raddi, Tim Marshall, Yunhui Ge, and Vincent A. Voelz. "Model Selection using Bayesian Inference of Conformational Populations with Replica-Averaging." J. Chem. Theory Comput. (2025), preprint; article
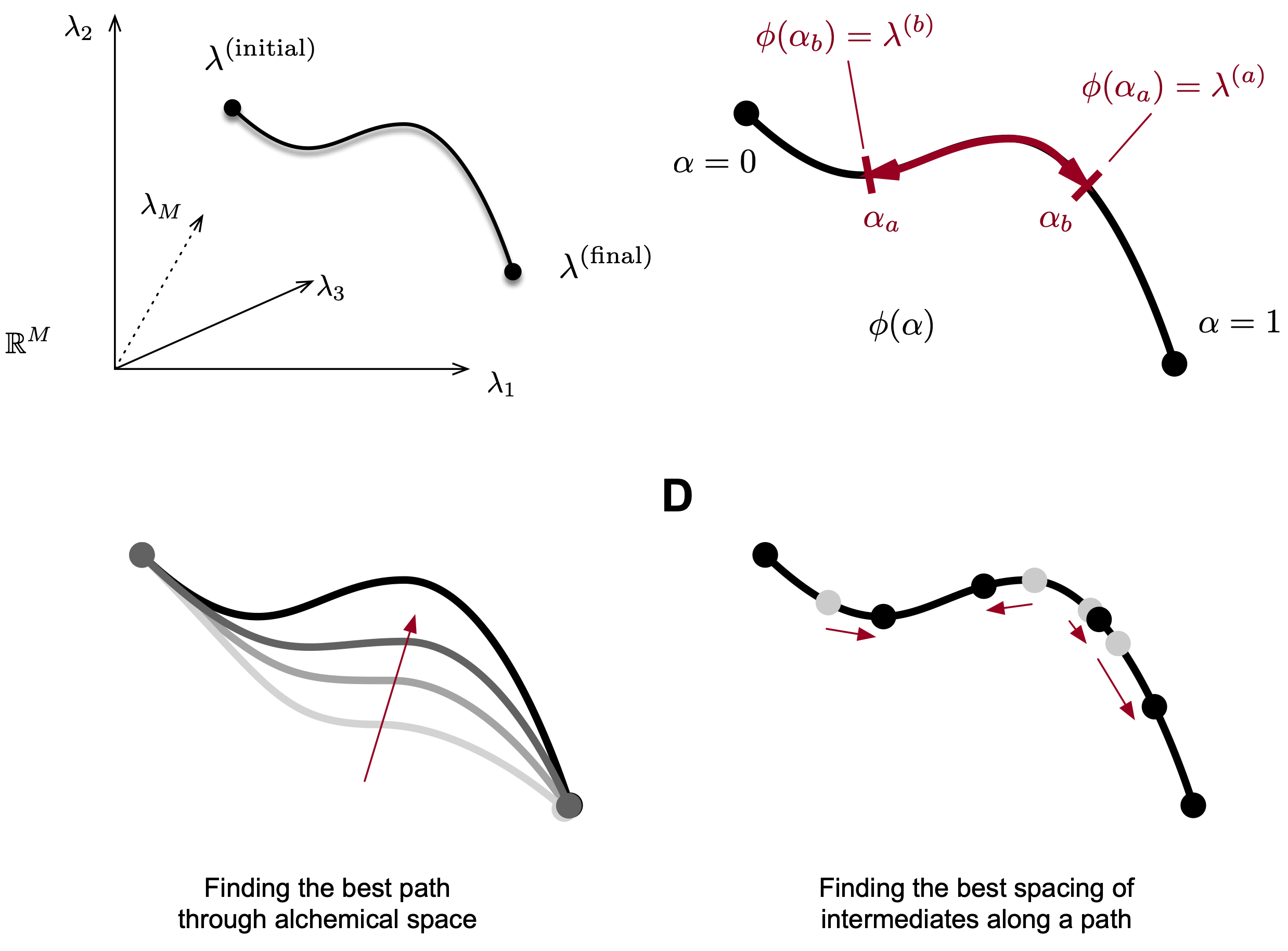
How can we efficiently choose and space alchemical intermediates in free energy simulations? This work introduces a simple method based on thermodynamic length and predicted mixing times to automate intermediate placement.
Dylan Novack, Robert M. Raddi, Si Zhang, Matthew F.D. Hurley and Vincent A. Voelz. "A simple method to optimize the spacing and number of alchemical intermediates in expanded ensemble free energy calculations." J. Chem. Inf. Model. (2025), preprint; article
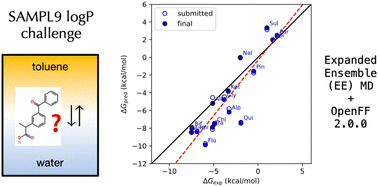
How accurately can expanded ensemble simulations predict logP values for water–toluene partitioning? This study benchmarks predictions against experimental results from the SAMPL9 blind challenge.
Steven Goold,Robert M. Raddi and Vincent A. Voelz. "Expanded Ensemble Predictions of Toluene‑Water Partition Coefficients in the SAMPL9 log$P$ Challenge." Phys. Chem. Chem. Phys., 2025, 27, 6005–6013 preprint; article

How well accurately can we predict absolute binding free energies using all-atom molecular dynamics simulations? In this work, we use the SAMPL9 community-wide blind host--guest challenge to test our expanded ensemble workflow for predicting absolute binding free energies for 13 small molecules against pillar[6]arene.
Matthew F.D. Hurley$^{\large\boldsymbol{\dagger}}$, Robert M. Raddi$^{\large\boldsymbol{\dagger}}$, Jason Pattis and Vincent A. Voelz. "Expanded Ensemble Predictions of Absolute Binding Free Energies in the SAMPL9 Host-Guest Challenge." Phys. Chem. Chem. Phys., 2023,25, 32393-32406. preprint; article
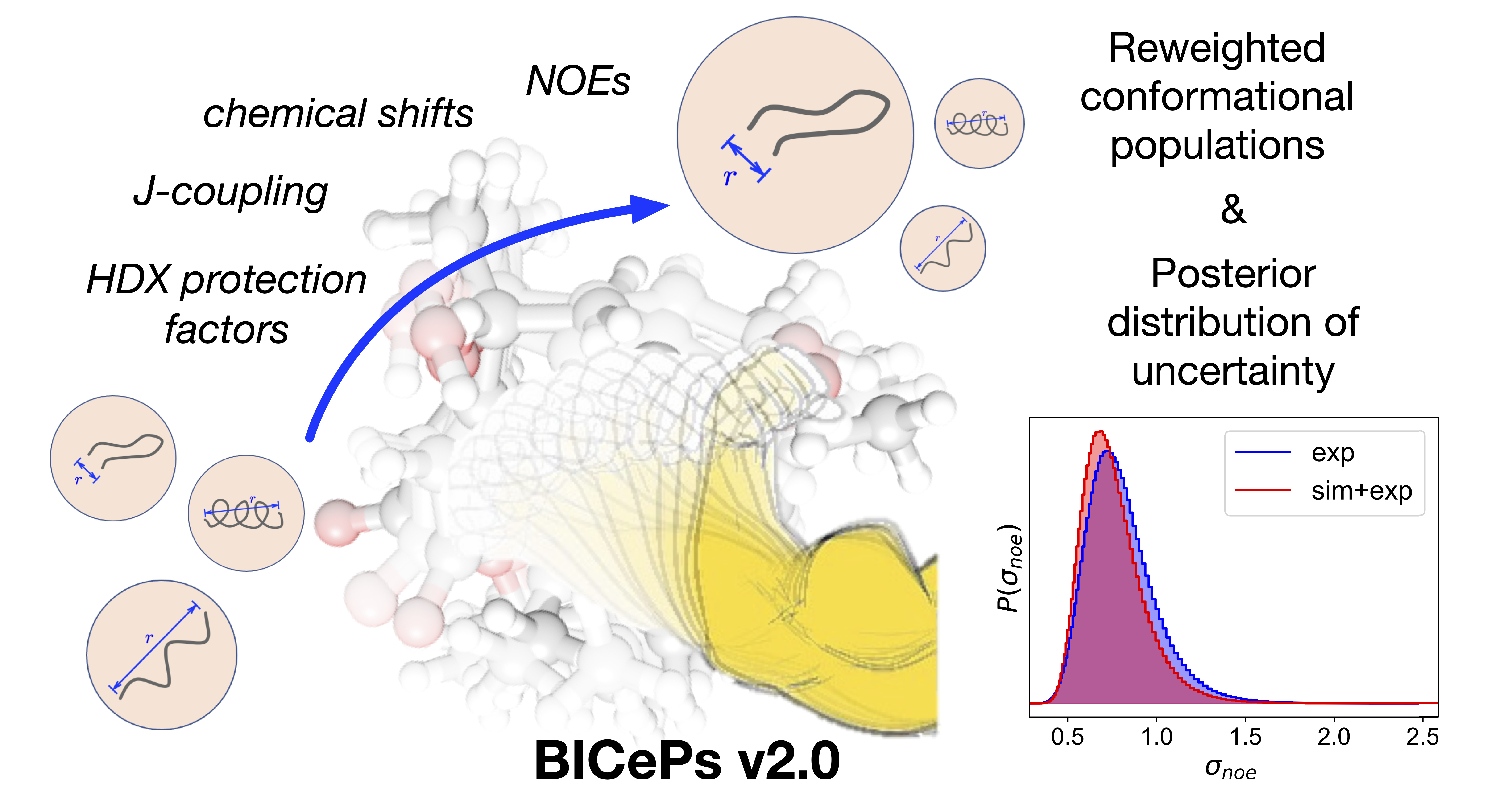
An easy-to-use Python package for structural ensemble refinement using ensemble-averaged experimental data.
Robert M. Raddi, Yunhui Ge, and Vincent A. Voelz. "BICePs v2.0: Software for Ensemble Reweighting using Bayesian Inference of Conformational Populations." J. Chem. Inf. Model. 2023, 63, 8, 2370–2381. preprint; article
